Nucleophilic-Substitution-7
If the E1cB mechanism is correct we should be able to recover labelled 2-phenylethyl bromide after a partial transformation to styrene. On the other hand, there should be no incorporation of deuterium if the E2 mechanism is operative. Actual experiments have shown that there is no deuterium incorporation and hence the E1cB mechanism does not operate in this case. However, this mechanism does operate under very special circumstances. 1,1,1 Trifluoro-2,2dichloroethane (III), due to strong C–F bond which take time to breakdown so partical negative charge develop on carbon atom, undergoes base-catalyzed exchange of -hydrogen atom with the solvent deuterium faster than dehydrofluorination.
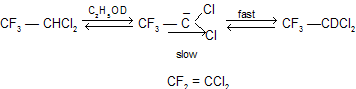
A strong carbon-fluorine bond (poor leaving ability of fluoride ion also) coupled with the electron withdrawing effect of the halogens explain the formation of carbanion before elimination.
(ii) E1 Reaction : the unimolecular mechanism
The main feature of this mechanism is that under the influence of solvation forces, the electron-attracting group (‘leaving group’) breaks away along with the bonding electrons. The resultant carbonium ion subsequently loses a proton to the solvent or to some other proton acceptor.
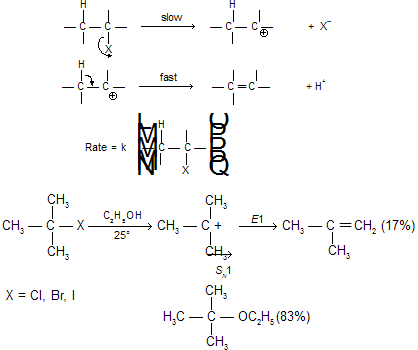
Acid-catalyzed dehydration of alcohols to alkenes provides another example of E1 reactions. The various steps can be written in the following way. Synthesis of carbocation occurs so molecular rearrangement is possible in differant reactions.
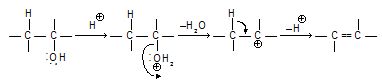
(iii) Orientation in elimination reactions
The elimination reactions of unsymmetrical substrates usually yield mixtures of all possible products. There are two empirical rules governing the orientation in these reactions.
a. The Saytzeff Rule
It states that neutral substrates (alkyl halides or sulphonates) capable of forming a double bond in either direction of the chain preferably yield that alkene in which there is greater number of alkyl groups attached to the double bond. This rule applies to E1 reactions and to most of E2 reactions. The following examples are typical:
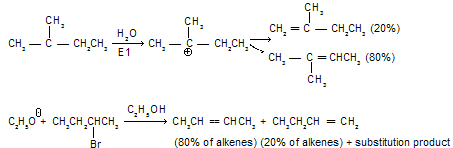
The preferential formation of more substituted alkenes in the above reactions can be correlated with the relative stability of various alkenes. Calculations from heats of combustion and hydrogenation establish that the stability of a double bond is increased by alkyl substitution on the double-bonded carbon atoms. Thus the stabilities of alkenes follow the order