Every Thing About 'Heat Capacity'
Definition of heat capacity
Heat capacity is mathematically defined as the ratio of a small amount of heat ?Q added to the body, to the corresponding small increase in its temperature dT:

For thermodynamic systems with more than one physical dimension, the above definition does not give a single, unique quantity unless a particular infinitesimal path through the system?s phase space has been defined (this means that one needs to know at all times where all parts of the system are, how much mass they have, and how fast they are moving). This information is used to account for different ways that heat can be stored as kinetic energy (energy of motion) and potential energy (energy stored in force fields), as an object expands or contracts. For all real systems, the path through these changes must be explicitly defined, since the value of heat capacity depends on which path from one temperature to another, is chosen. Of particular usefulness in this context are the values of heat capacity for constant volume, CV, and constant pressure, CP. These will be defined below.
Heat capacity of compressible bodies
The state of a simple compressible body with fixed mass is described by two thermodynamic parameters such as temperature T and pressure p. Therefore as mentioned above, one may distinguish between heat capacity at constant volume, CV, and heat capacity at constant pressure, Cp:

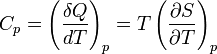
where
- ?Q is the infinitesimal amount of heat added,
- dT is the subsequent rise in temperature.
The increment of internal energy is the heat added and the work added:

So the heat capacity at constant volume is

The enthalpy is defined by H = U + PV. The increment of enthalpy is
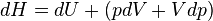
which, after replacing dU with the equation above and cancelling the PdV terms reduces to:

So the heat capacity at constant pressure is

Note that this last ?definition? is a bit circular, since the concept of ?enthalpy? itself was invented to be a measure of heat absorbed or produced at constant pressures (the conditions in which chemists usually work). As such, enthalpy merely accounts for the extra heat which is produced or absorbed by pressure-volume work at constant pressure. Thus, it is not surprising that constant-pressure heat capacities may be defined in terms of enthalpy, since ?enthalpy? was defined in the first place to make this so.
Relation between specific heats
Measuring the heat capacity at constant volume can be prohibitively difficult for liquids and solids. That is, small temperature changes typically require large pressures to maintain a liquid or solid at constant volume implying the containing vessel must be nearly rigid or at least very strong (see coefficient of thermal expansion and compressibility). Instead it is easier to measure the heat capacity at constant pressure (allowing the material to expand or contract as it wishes) and solving for the heat capacity at constant pressure using mathematical relationships derived from the basic thermodynamic laws. Starting from the combined law of thermodynamics one can show,

where,
- ? is the coefficient of thermal expansion, and
- ?T is the isothermal compressibility.
For an ideal gas this reduces to the simple relation,

Specific heat capacity
The specific heat capacity of a material is

which in the absence of phase transitions is equivalent to

where
- C is the heat capacity of a body made of the material in question,
- m is the mass of the body,
- V is the volume of the body, and
 is the density of the material.
is the density of the material.
For gases, and also for other materials under high pressures, there is need to distinguish between different boundary conditions for the processes under consideration (since values differ significantly between different conditions). Typical processes for which a heat capacity may be defined include isobaric (constant pressure, dp = 0) or isochoric (constant volume, dV = 0) processes. The corresponding specific heat capacities are expressed as


A related parameter to c is  , the volumetric heat capacity. In engineering practice,
, the volumetric heat capacity. In engineering practice,  for solids or liquids often signifies a volumetric heat capacity, rather than a constant-volume one. In such cases, the mass-specific heat capacity (specific heat) is often explicitly written with the subscript m, as
for solids or liquids often signifies a volumetric heat capacity, rather than a constant-volume one. In such cases, the mass-specific heat capacity (specific heat) is often explicitly written with the subscript m, as  . Of course, from the above relationships, for solids one writes
. Of course, from the above relationships, for solids one writes
, the volumetric heat capacity. In engineering practice, for solids or liquids often signifies a volumetric heat capacity, rather than a constant-volume one. In such cases, the mass-specific heat capacity (specific heat) is often explicitly written with the subscript m, as . Of course, from the above relationships, for solids one writes
Dimensionless heat capacity
The dimensionless heat capacity of a material is

where
- C is the heat capacity of a body made of the material in question (J·K?1)
- n is the amount of matter in the body (mol)
- R is the gas constant (J·K?1·mol?1)
- nR=Nk is the amount of matter in the body (J·K?1)
- N is the number of molecules in the body. (dimensionless)
- k is Boltzmann?s constant (J·K?1·molecule?1)
Again, SI units shown for example.
Theoretical models
Gas phase
The specific heat of the gas is best conceptualized in terms of the degrees of freedom of an individual molecule. The different degrees of freedom correspond to the different ways in which the molecule may store energy. The molecule may store energy in its translational motion according to the familiar formula

where m is the mass of the molecule and [vx,vy,vz] is velocity of the center of mass of the molecule. Each direction of motion constitutes a degree of freedom, so that there are three translational degrees of freedom.
In addition, a molecule may have rotational motion. The kinetic energy of rotational motion is generally expressed as
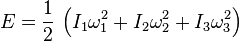
where I is the moment of inertia tensor of the molecule, and [?1,?2,?3] is the angular velocity pseudovector (in a coordinate system aligned with the principle axes of the molecule). In general, then, there will be three additional degrees of freedom corresponding to the rotational motion of the molecule, (For linear molecules one of the inertia tensor terms vanishes and there are only two rotational degrees of freedom). The degrees of freedom corresponding to translations and rotations are called the ?rigid? degrees of freedom, since they do not involve any deformation of the molecule.
The motions of the atoms in a molecule which are not part of its gross translational motion or rotation may be classified as vibrational motions. It can be shown that if there are n atoms in the molecule, there will be as many as 3n ? 3 ? nr vibrational degrees of freedom, where nr is the number of rotational degrees of freedom. The actual number may be less due to various symmetries.
If the molecule could be entirely described using classical mechanics, then we could use the theorem of equipartition of energy to predict that each degree of freedom would have an average energy in the amount of (1/2)kT where k is Boltzmann?s constant and T is the temperature. Our calculation of the heat content would be straightforward. Each molecule would be holding, on average, an energy of (f/2)kT where f is the total number of degrees of freedom in the molecule. The total internal energy of the gas would be (f/2)NkT where N is the total number of molecules. The heat capacity (at constant volume) would then be a constant (f/2)Nk , the specific heat capacity would be (f/2)k and the dimensionless heat capacity would be just f/2.
The various degrees of freedom cannot generally be considered to obey classical mechanics. Classically, the energy residing in each degree of freedom is assumed to be continuous - it can take on any positive value, depending on the temperature. In reality, the amount of energy that may reside in a particular degree of freedom is quantized: It may only be increased and decreased in finite amounts. A good estimate of the size of this minimum amount is the energy of the first excited state of that degree of freedom above its ground state. For example, the first vibrational state of the HCl molecule has an energy of about 5.74 × 10?20 joule. If this amount of energy were deposited in a classical degree of freedom, it would correspond to a temperature of about 4156 K.
If the temperature of the substance is so low that the equipartition energy of (1/2)kT is much smaller than this excitation energy, then there will be little or no energy in this degree of freedom. This degree of freedom is then said to be ?frozen out". As mentioned above, the temperature corresponding to the first excited vibrational state of HCl is about 4156 K. For temperatures well below this value, the vibrational degrees of freedom of the HCL molecule will be frozen out. They will contain little energy and will not contribute to the heat content of the HCl gas.
It can be seen that for each degree of freedom there is a critical temperature at which the degree of freedom ?unfreezes? and begins to accept energy in a classical way. In the case of translational degrees of freedom, this temperature is that temperature at which the thermal wavelength of the molecules is roughly equal to the size of the container. For a container of macroscopic size (e.g. 10 cm) this temperature is extremely small and has no significance, since the gas will certainly liquify or freeze before this low temperature is reached. For any real gas we may consider translational degrees of freedom to always be classical and contain an average energy of (3/2)kT per molecule.
The rotational degrees of freedom are the next to ?unfreeze". In a diatomic gas, for example, the critical temperature for this transition is usually a few tens of kelvins. Finally, the vibrational degrees of freedom are generally the last to unfreeze. As an example, for diatomic gases, the critical temperature for the vibrational motion is usually a few thousands of kelvins.
It should be noted that it has been assumed that atoms have no rotational or internal degrees of freedom. This is in fact untrue. For example, atomic electrons can exist in excited states and even the atomic nucleus can have excited states as well. Each of these internal degrees of freedom are assumed to be frozen out due to their relatively high excitation energy. Nevertheless, for sufficiently high temperatures, these degrees of freedom cannot be ignored.
Monatomic gas
In the case of a monatomic gas such as helium under constant volume, if it assumed that no electronic or nuclear quantum excitations occur, each atom in the gas has only 3 degrees of freedom, all of a translational type. No energy dependence is associated with the degrees of freedom which define the position of the atoms. While, in fact, the degrees of freedom corresponding to the momenta of the atoms are quadratic, and thus contribute to the heat capacity. There are N atoms, each of which has 3 components of momentum, which leads to 3N total degrees of freedom. This gives:


where
- CV is the heat capacity at constant volume of the gas
- CV,m is the molar heat capacity at constant volume of the gas
- N is the total number of atoms present in the container
- n is the number of moles of atoms present in the container (n is the ratio of N and Avogadro?s number)
- R is the ideal gas constant, (8.314570[70] J K?1mol?1). R is equal to the product of Boltzmann?s constant kB and Avogadro's number